HVAC systems play a central role in maintaining compliance within biopharmaceutical facilities, making such systems essential during renovation projects. Because these systems are often deeply integrated into existing infrastructure, the design service provider should become familiar with all aspects of the facility’s existing HVAC system to accurately define the scope and scale of required modifications to this critical mechanical system.
At this stage, project teams should document key HVAC design criteria, including:
- HVAC area classifications
- Air pressure relationships and differentials
- Air-handling unit boundaries and capacities
- Local exhaust requirements
- Humidity control requirements
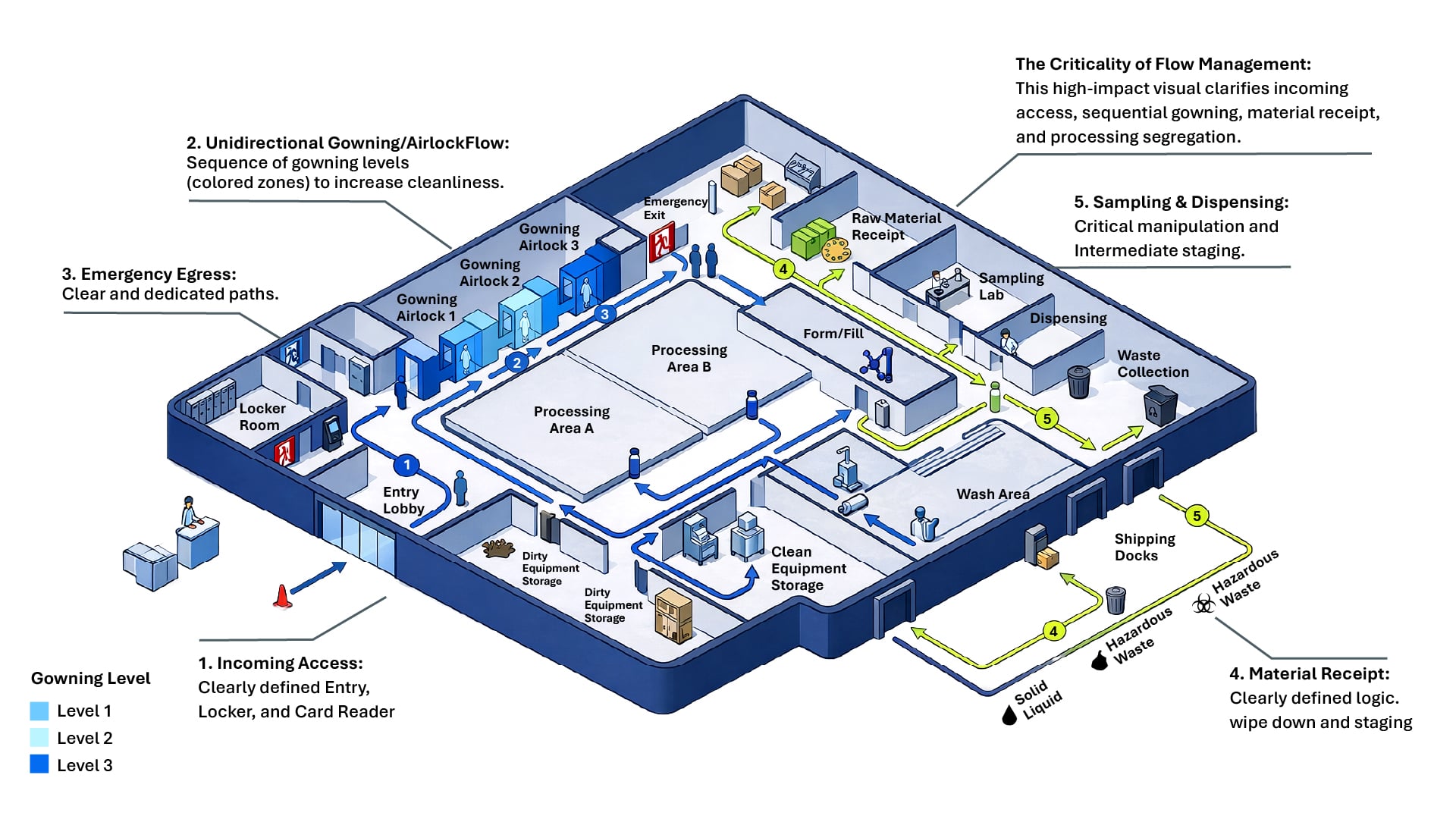
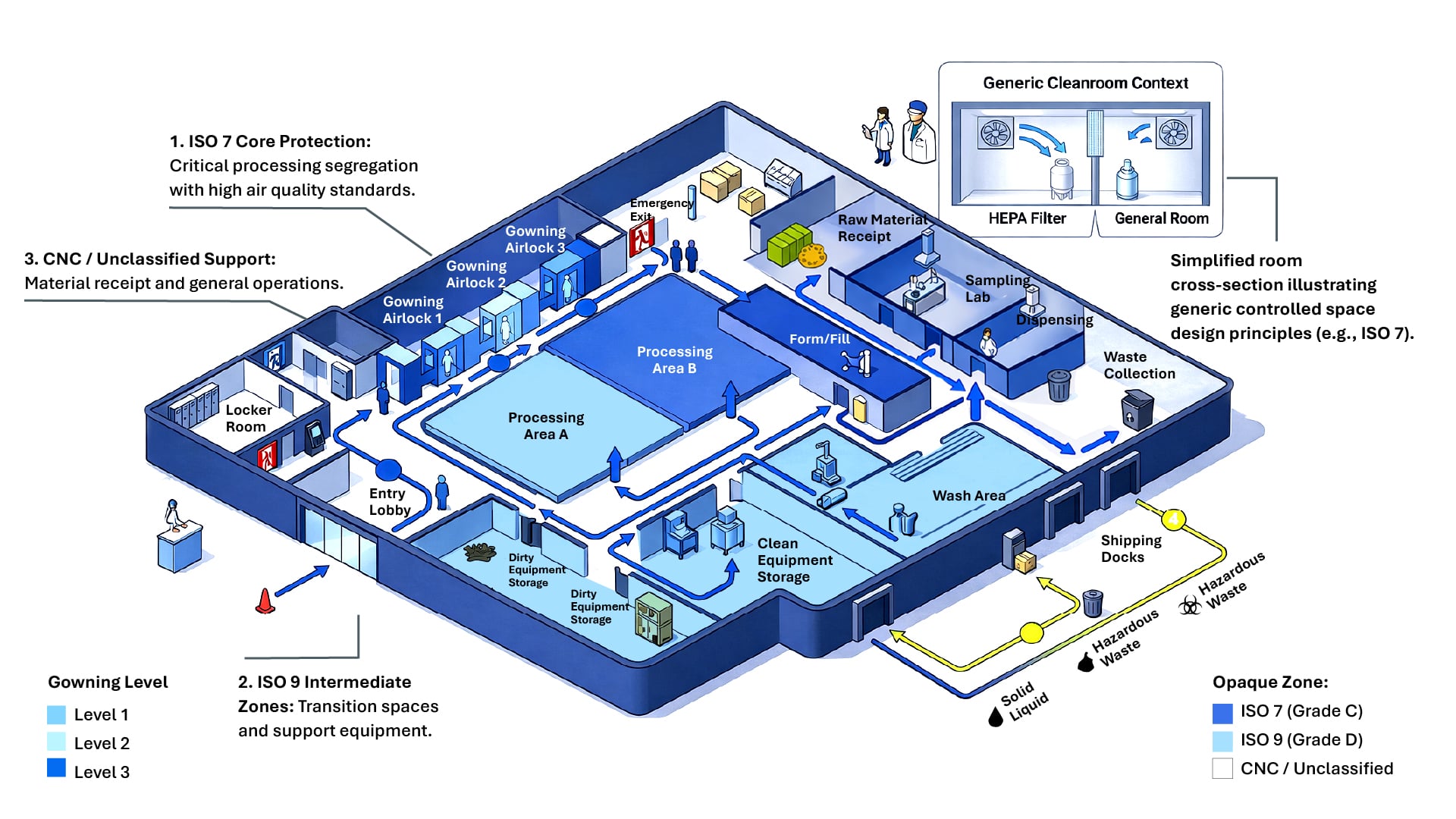
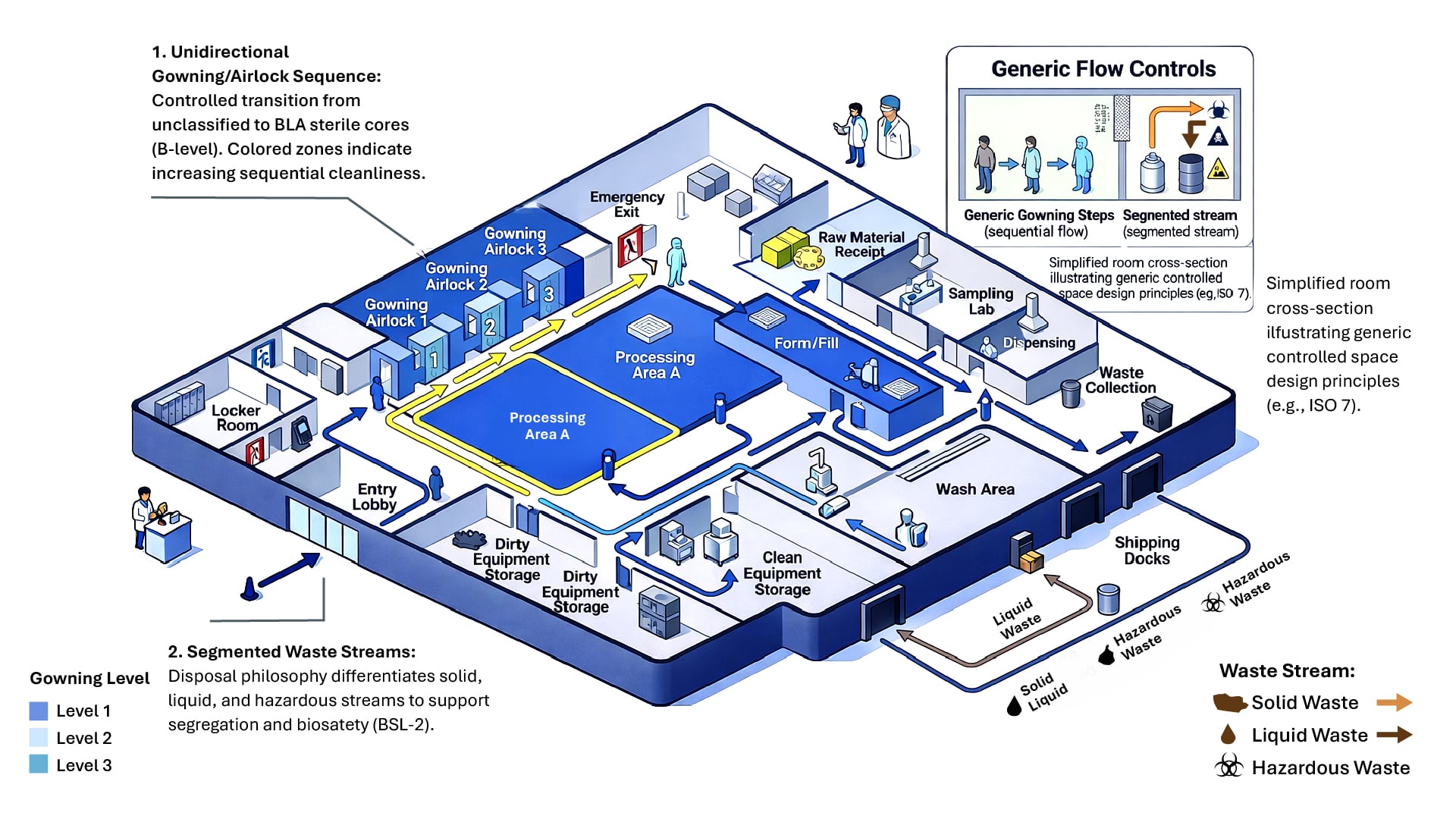
HVAC area classifications — defined by the number of particles 0.5 microns and larger per cubic foot of air — should support current Good Manufacturing Practices (cGMPs), which have evolved over time. These classifications are often most effectively communicated through visual overlays on general arrangement drawings, allowing teams to quickly understand how different areas relate to one another.
In addition, HEPA filter locations should be clearly documented, whether in-line or terminal. These decisions can carry regulatory implications, particularly for facilities operating in multiple global markets.
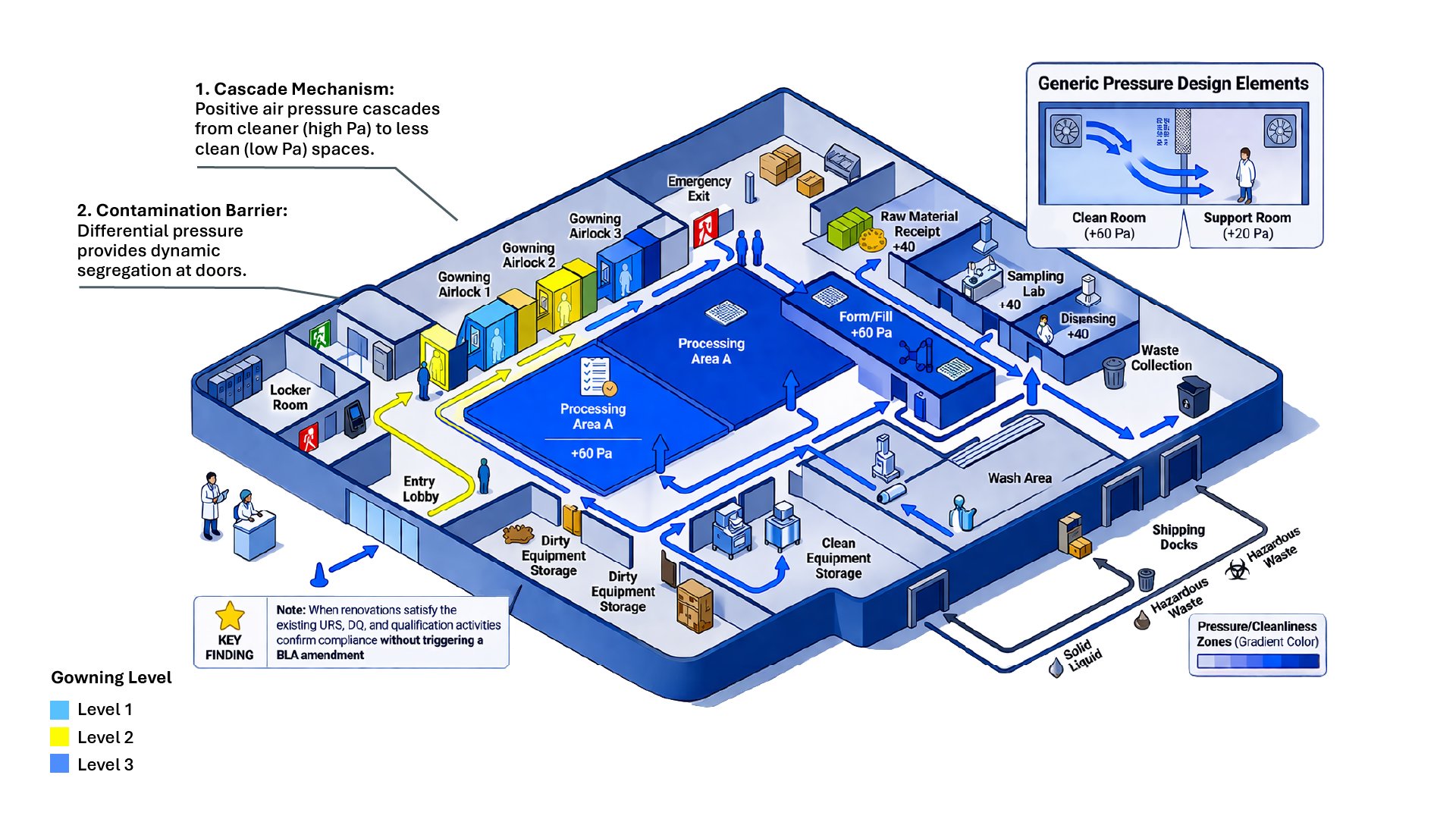
Air pressure differentials are typically designed to cascade from cleaner areas to less controlled spaces, helping maintain proper containment and product protection. Exceptions may apply in cases where containment is required for potent compounds, steroids or specific host cell organisms that fall outside standard Good Large-Scale Practice definitions.
Air-handling unit (AHU) boundaries can be overlaid on the facility floor plan to show which areas each AHU serves. This analysis helps determine whether an existing unit could support the renovated space or if a new unit or once-through air would be required.
Mapping AHU boundaries across the facility helps identify which systems serve each area and whether existing units could support the renovated space. In some cases, this analysis may indicate the need for new equipment or once-through air systems.
Local exhaust requirements should also be documented to aid the HVAC design for the area in question as they can adversely impact room pressurizations and balancing activities if not planned for in advance.
Approaches to area classification have varied historically, even for similar operations. For example, buffer preparation in licensed biologics facilities has ranged from less stringent environments, such as Grade D or ISO 9, to more tightly controlled spaces like Grade B or ISO 7. These variations are often the result of previously unclear regulatory guidance.
Recent updates have introduced more flexibility, allowing organizations to reduce capital and operating costs while still meeting regulatory expectations. Guidance from the International Council for Harmonization (ICH Q7) and the International Society for Pharmaceutical Engineering (ISPE) supports the use of controlled, nonclassified (CNC) spaces in certain scenarios, particularly when processes are closed. In these cases, CNC environments may be appropriate for activities such as nonsterile media and buffer preparation when solutions are later sterile-filtered.
As these options are evaluated, alignment with the client organization remains important. Decisions around classification and system design should reflect both regulatory expectations and the organization’s risk tolerance before being incorporated into the final facility design.
Other factors that tend to have a significant impact to the facility layout from a cGMP and regulatory compliance standpoint include:
- Classification and perceived risk of the host cell organism in biologics facilities (i.e., whether the organism falls under Good Large-Scale Practice or biosafety level 2)
- Segregation considerations (i.e., pre- and post-viral inactivation, live versus inactivated cells)
- Potential markets for the drug substance being produced